
What is Primary Ciliary Dyskinesia?
Primary Ciliary Dyskinesia (PCD) is a clinically and genetically heterogeneous group of congenital diseases based on the malfunction of motile cilia. Approximately half of those affected with PCD present a mirror-image arrangement of the internal organs (situs inversus), which is referred as Kartagener syndrome. An estimated 500,000 people worldwide are affected by PCD, but unfortunately the diagnosis is made in only a small proportion of patients. This is because the disease is unknown even to many doctors. Moreover, the tools necessary for the diagnosis of PCD requires expertise and is time consuming and expensive; thus it is currently available only in a few specialized centers.
What are cilia and the importance they have in PCD?
Cilia are thin hair-like constituents of virtually every cell in the human body. The airways of the upper respiratory tract (nose, paranasal sinuses) including the smallest bronchi of the lungs are lined with billions of moving cilia. A coordinated movement of the cilia serves an important transport function and thus contributes to the cleaning of the airways. People with PCD have no functioning cilia. As a result, mucus progressively accumulates in the airways, which cannot be removed. As a result, PCD patients suffer frequent infections of the lungs, middle ear and sinuses. Over time, these infections become chronic and can lead to permanent damage, especially in the lungs.
What are the symptoms of PCD?
The symptoms of PCD can be already seen at birth: breathing is hard, the nose is obstructed by secretions, coughing is present, and part of the lung may even be collapsed. In addition, PCD patients develop a chronic cough and frequently have clinically severe respiratory infections. Since airway cleaning is disturbed, not only the respiratory tract in the lung but also the nasal passages have chronic obstruction. Already in early infancy, there is an enlargement of the tonsils (adenoids) and there are frequent middle ear infections. Often in these patients, the adenoids are removed and timpani tubes are placed to facilitate clearance - if symptoms do not improve, chronic otitis media may result. Chronic middle ear effusions are a frequent cause of hearing loss, which is often spontaneous in adolescensce. School age children suffer from frequent sinus infections. The course of the disease is usually determined by the extent of pulmonary involvement. Infections of the lung may lead to collapse of a portion of the lung; if it is not possible to reopen this section of the lung, a chronic infection may develop there. Additionally, thick mucus can completely obstruct the bronchi and lead to destruction of not only the bronchi (bronchiectasis) but also the entire lung section. Since these damaged sections of the lung are very susceptible to pathogens, infections are relatively unhindered and can spread to other parts of the lung.
Importantly, the care of patients with PCD varies with the course of the disease for each individual. While some patients are diagnosed as newborns, others may manifest symptoms only in infancy or even later. Similarly, the onset and progression of chronic lung disease is quite variable and can influence measures taken for therapy.
Which organs are affected?
Since cilia occur in almost all cells of the body, other organ systems can be affected. Male PCD patients are often infertile due to a lack of sperm motility. This is because sperm tails, which are used to propel sperm cells, are either dysmotile or immotile. Even the female fallopian tubes are lined with motile cilia. The functional role of these cilia is still not fully understood. Potentially affected women have a slightly increased risk for ectopic pregnancies. Rarely, other diseases such as hydrocephalus (enlargement of the cerebrospinal fluid spaces), retinitis pigmentosa (a rare retinal disease), cystic kidney disease, certain heart defects or hearing loss also occur in PCD patients. The phenomenon of situs inversus (mirror-image reversal of the arrangement of the internal organs) is explained by the fact that it results from ciliary dysfunction in the area of the so-called "embryonic node" during embryonic development, which normally dictates the proper left-right distribution of organs. This does not lead to impairment of organ function, so situs inversus plays no additional clinical significance.
For a review article with more information, please see the bottom of the page for a downloadable document.
How is PCD diagnosed?
The diagnosis of PCD is complex, expensive and dependent on the experience of the examiner. In the preliminary diagnosis, the nitric oxide (NO) concentration can be determined in the nose. Patients with PCD have a significantly reduced nasal NO concentration. However, the significance of this diagnosis is not entirely clear at present. Low nasal NO concentration alone is certainly not proof for the diagnosis of PCD. Further information can be found in the following link: information nasal NO. In almost all cases, a diagnosis by analysis of ciliated cells of the nasal mucosa is possible. For this purpose, a soft brush is used to remove ciliated cells present in the nose. This is quick (20 seconds) and can be harmlessly applied to neonates and infants. Only in exceptional cases, the removal of tissue from the bronchial tubes is required. This usually happens during a bronchoscopy, which is necessary to clear the airways when the patient has difficulty breathing. The diagnosis of PCD is based on three clinically accepted methods, in accordance with European directives (see download "consensus statement" on the bottom). Currently, our hospital is the world's only center that uses all three methods for a diagnosis of PCD:
I. First, cells directly derived from the airway are analysed for ciliary beating by high-frequency video microscopy. Here the experienced investigators know with great certainty whether PCD exists or not. In the most common cause of PCD, in which loss of the outer dynein arms (ODA) are discernible ultrastructurally, the cilia are immotile or show only slight twitches. DNAH5 is a component of the ODA, and mutations in the corresponding gene usually lead to the loss of the ODA. Other changes are harder to detect because they modify the movement (e.g., rigid or hyperkinetic ciliary beating).
Respiratory epithelial cells of healthy volunteers.
Respiratory epithelial cells from a PCD patient with DNAH5 mutations.
Diagnostic immunofluorescence microscopy:

IF staining of healthy, respiratory epithelial cells. The nucleus is blue, microtubules of the cilia are green, and the outer dynein arms (ODA) are colored red. The yellow color indicates the overlap of green and red signals.

IF staining of a patient with an ODA defect: Along the cilium is no red (ODA) staining visible, only Tubulin (green) is shown.
II. For further clarification, a novel diagnostic immunofluorescence microscopy (IF) method developed by our laboratory can be used to help identify individual defects of ciliary structure.
More information can be found on the "IF-diagnostics" page.
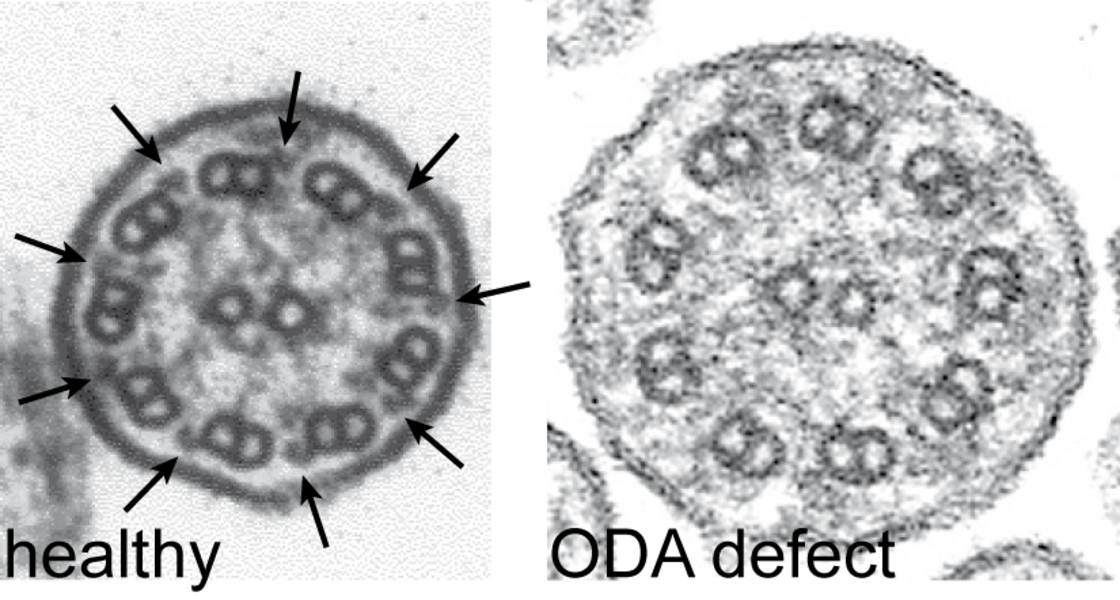
Cilia cross sections from healthy volunteers show nine peripheral pairs of microtubuli with outer (arrows left) and inner dynein arms. PCD patients with mutations in DNAH5 exhibit an ODA defect.
III. In addition, electron microscopy of the cilia can also be performed to illustrate defects of ciliary structure. Most common are defects of the outer and inner dynein arms.
The dynein arms are large protein complexes that consist of heavy, medium and light dynein chains and other associated proteins.
Mutations in the corresponding genes, such as DNAH5 (coding for a heavy dynein chain), result in the loss of the entire outer dynein arms.
In addition, we offer all patients the opportunity to participate in our study to investigate the molecular basis of primary ciliary dyskinesia and to establish a genetic diagnosis. For more information, see the link below to "Genetic of PCD". In recent years, our group has been able to demonstrate in many patients the changes in the genetic information (mutations) of genes (including DNAH5, DNAH11, DNAI1, DNAI2, LRRC50, KTU, CCDC39, CCDC40, CCDC103, CCDC164, DYX1C1, HYDIN and ARMC4).
The genetic screening of known PCD mutations are carried out in collaboration with the Institute for Human Genetics at the University Hospital of Muenster (Univ.-Prof. Dr. P. Wieacker). Before submitting samples to our laboratory for genetic analysis, we ask you to complete a transfer certificate.
If you are interested in comprehensive information on the genetics of PCD, please visit "Genetic of PCD".
How is PCD treated?
Without the earliest possible diagnosis and treatment, there is risk for permanent damage to the lungs, ears and sinuses. Although recurrent respiratory infections cannot be avoided completely by a consistent treatment, the frequency and severity of exacerbations can be reduced significantly. This intensive treatment includes measures to address impaired mucus transport. These include both a regular, intensive physiotherapy and inhalation therapy to liquefy and increase mobilization of mucus in the lungs. Additional measures to relieve mucus obstruction in the upper respiratory tract include regular rinsings or nasal inhalation via the paranasal sinuses. In addition, bacterial infections are immediately treated with antibiotics. If a section of the lung should collapse, it is necessary to determine by bronchoscopy whether the lung section can be preserved by bronchoscopy. If disease progression is marked by colonization of the airways with multi-resistant bacteria, often an inhaled antibiotic maintenance therapy is indicated. If this therapy does not stabilize the infection, the risk of chronic lung disease and lung failure is possible and the option of a lung transplant should be discussed.
In addition to respiratory disease, there is also a need for counseling or therapy for many patients: psychological, social, and legal advice if often needed to cope with the emotional and financial consequences of this disease. To address potential fertility problems in men, an andrological examination is useful to check whether the wish to father children can be fulfilled by means of artificial insemination. Other complaints such as heart defects, deafness and renal malformations, which may be present in a small proportion of those affected, require an appropriate medical professional interdisciplinary co-treatment.
In our pediatric pulmonology clinic (see link below), we support and advise patients of all age groups, from neonates to adults.
Link to Pediatric Pulmonology Clinics, Clinic of General Pediatrics at the University Hospital Münster
Links to additional pages of the homepage
Further readings
